
I don’t have much detail to answer the first question: he was an 18th century English mathematician who wrote something about probability, that was published after he died. That publication described something called Bayes’ theorem which is a way of incorporating the prior probability of something happening with the evaluation of new data to arrive at an updated probability. (I think) (someone tell me if I am way off base….)
So if you can calculate the probability of something, then you take into account any new information that you find, and recalculate a new probability. In some ways this is how we operate all the time in daily life, but Bayes thought of new ways of doing the calculations.
Anyway, I have heard about incorporating Bayesian probability into clinical trial design for a while, but I don’t recall having seen many examples. The idea being that the usual way of doing a trial is to assume that the two arms of the trial have an equal probability of being preferable (which is sort of like the null hypothesis) then do the trial with a specified sample size, avoid looking at the data until the end of the trial (if possible, but with safeguards built-in, just in case) and then do a test of significance and declare that one arm of the trial was better, hence the benefit of treatment B is proven. A Bayesian trial explicitly incorporates prior probability into the design, encourages adaptive trial designs with flexible sample sizes, encourages repeated looks at the data as they are accumulating, and at the end produces a new posterior probability that treatment B is better than treatment A.
Which all sounds interesting, and I know there are examples of it actually being done, but I wasn’t aware of any perinatal trials.
Here though is a trial of antenatal intervention for urinary tract obstruction called the PLUTO trial. It was a multicenter RCT with a planned sample size of 150 women and their fetuses. After several years they were only able to randomize 31 mothers, so they had to stop the trial, as they probably ran out of both money and patience. Now as far as I can tell the trial team did not put anything about Bayesian analysis into the registration documents, nor the published protocol, but, given that the sample size was so small and the results therefore rather negative, they proceeded with an analysis using Bayes’ methods. They used some estimates of what they thought, before the trial, was the probability that antenatal shunting would be the better treatment, and then calculated the new probability that shunting is better by adding in the new data.
So what did they find in the trial? Seven of the 16 babies randomized to be shunted survived to one year of age; and 3 of the 15 randomized to be treated conservatively, with evaluation and treatment after birth, survived.
That shows what a bad condition this is, the fetuses were eligible in cases of visualisation of an enlarged bladder and dilated proximal urethra, bilateral or unilateral hydronephrosis, and cystic parenchymal renal disease, if the obstetrician was unsure of the best clinical management.
The CONSORT diagram below shows you how horrendously complicated it is to do and then analyze a trial like this.
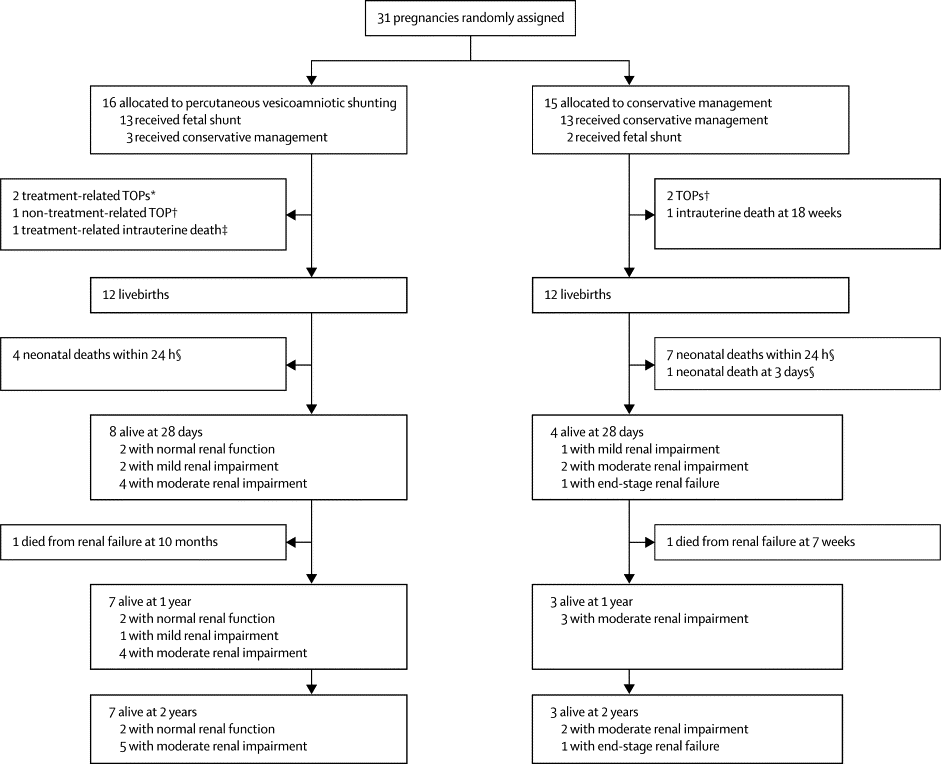
So of the 16 allocated to shunting, 3 were not shunted, and 1 changed their mind and decided to terminate the pregnancy, and there were 3 treatment related pregnancy losses. Some of those allocated to conservative treatment got shunted anyway, and some others terminated. So how do you decide whether shunting was better or not? I know the ‘correct answer’ is an ITT analysis, you just calculate according to the numbers randomized into each group. But I think there is a good case to be made here for, at least, taking out of the analysis the non-procedure related terminations, which gives you 7/15 vs 3/13 survivors to 2 years. I think an analysis by procedure actually performed is interesting also, but you always have to be very careful, as you don’t know why the protocol violations occurred, it may because of clinical factors that might also influence prognosis. Anyway the ‘as-treated’ analysis shows 8/14 survivors who were shunted vs 2/14 conservative.
This is suggestive that maybe the shunting really did help, but it is clearly still a maybe.
The reasons for going into so much detail of this trial (apart from the fact that it is a trial that we really needed, and it is a great shame that they were unable to get a bigger sample) is that the authors then incorporate a Bayesian analysis, they determine a prior probability that shunting would be better, then add in the new results, and then calculate a new probability that shunting really improves survival. They calculate the prior probability as being 0.79 that shunting is preferable, and with the new trial data they state that the posterior probability is now 0.86.
So this is a Bayesian analysis of results a trial that was planned as a more conventional trial to determine superiority. My major problem with the analysis in this trial is that the prior probability is based on asking ‘experts’ what they thought. I think that even prior probabilities should be based on some sort of data, now having said that the prior observational data were actually more positive than the experts’ opinions, which just shows you how careful you have to be about observational data, it can be seriously biased.
Trials actually planned using Bayesian methods are also interesting. I know little about this, so I was pleased to find this document, it is a document reproducing what was taught to participants in a workshop on clinical research methods. It is a great introduction to what clinical research is all about, and there is quite a long and detailed section about Bayesian trial design.
I have been wondering about trials like the future lactoferrin trials, for example, if we try to calculate the likelihood that lactoferrin will prove to be very potent at preventing nosocomial infections (for which we actually have some hard data), and incorporate that into trial design, perhaps we can reduce the required sample size for future trials, and get an answer sooner.
By the way all the articles about Bayes use the same image, which probably isn’t him!








